現在(2026)的基因定序以次世代定序(Next Generation Sequencing, NGS;有別於 Sanger sequencing)為主。醫生參與的部份多為變異判讀(Variant Interpretation)並產出正式報告,所拿到的資料也已經經過對齊(Alignment)、變異偵測(Variant Calling)與變異註解(Variant Annotation)。
變異判讀(Variant Interpretation)依循美國醫學遺傳學暨基因體學學會(ACMG)的指引。每位醫師的作法不同,我自己則切分為幾個步驟:臨床表型整理(Clinical Phenotype Processing)→ 候選基因變異篩選定位(Candidate Genetic Variant Selections) → 致病力查詢(Pathogenicity Queries)→ 臨床意義未明的變異評估(Variants of Uncertain Significance Evaluation)→ 基因型與表型對照(Genotype-Phenotype Mapping) 。以下將分段說明。
臨床表型整理(Clinical Phenotype Processing)
這個部份非常需要臨床醫師的經驗,甚至需要專科或次專科醫師才有辦法對病人的表型有著細緻地掌握,並整理成可供檢索的語彙。以目前 (2026) 的科技與制度而言,直接用機器爬往往會 GIGO。癌症則可以靠「病理診斷」來找尋其因應的常見基因變異,至於罕見先天性疾病,往往需要兒童遺傳科甚至不同的專科一起協助找出異常之處。
表型可以先區分為多系統(multi-system disease)或單系統(single system disease)。雖然像地中海型貧血有多系統的影響(比如說塞到各器官的血栓、因為長期輸血而併發的鐵質沉積症),它最核心的問題還是在血球中的血紅素變異上,因此還是傾向將它列為單系統疾病並設計因應的常見異常基因組套。
罕見疾病的表現型可以透過 Human Phenotype Ontology (HPO) 進行查找。臨床表現與疾病之間千絲萬縷的關連一時半刻無法說清楚,它牽涉許多認知論對於物事的切分,有興趣者歡迎再延伸閱讀以下數篇:
〈所謂診斷〉(Wu, 2017)
〈疾病的本質〉(Wu, 2018)
〈醫學知識論 Medical Epistemology:臨床知識歸位與框架消融〉(Wu, 2024)
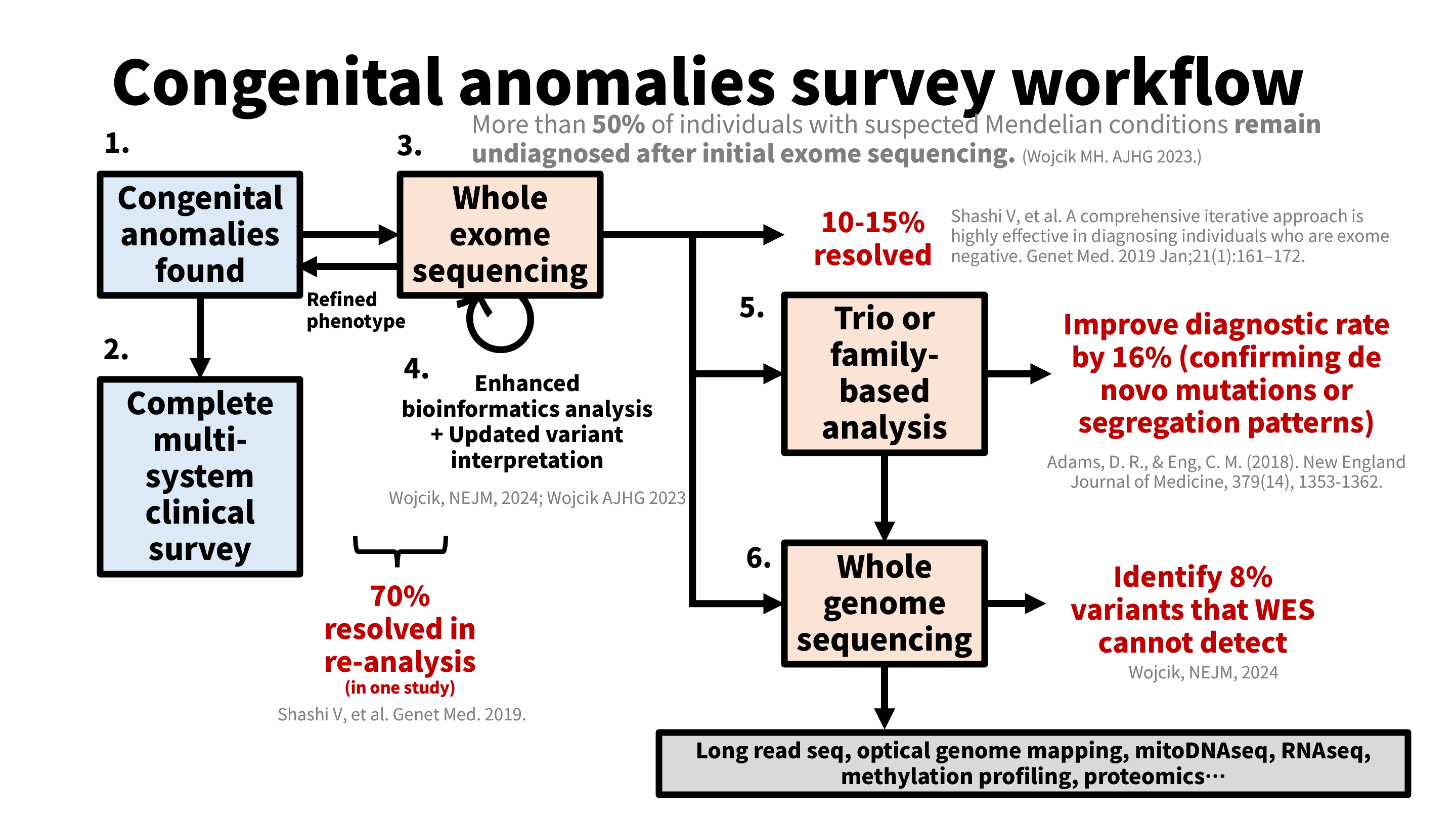
有許多資料庫整合臨床表型與基因型的關聯性供我們檢索,用下圖標示。
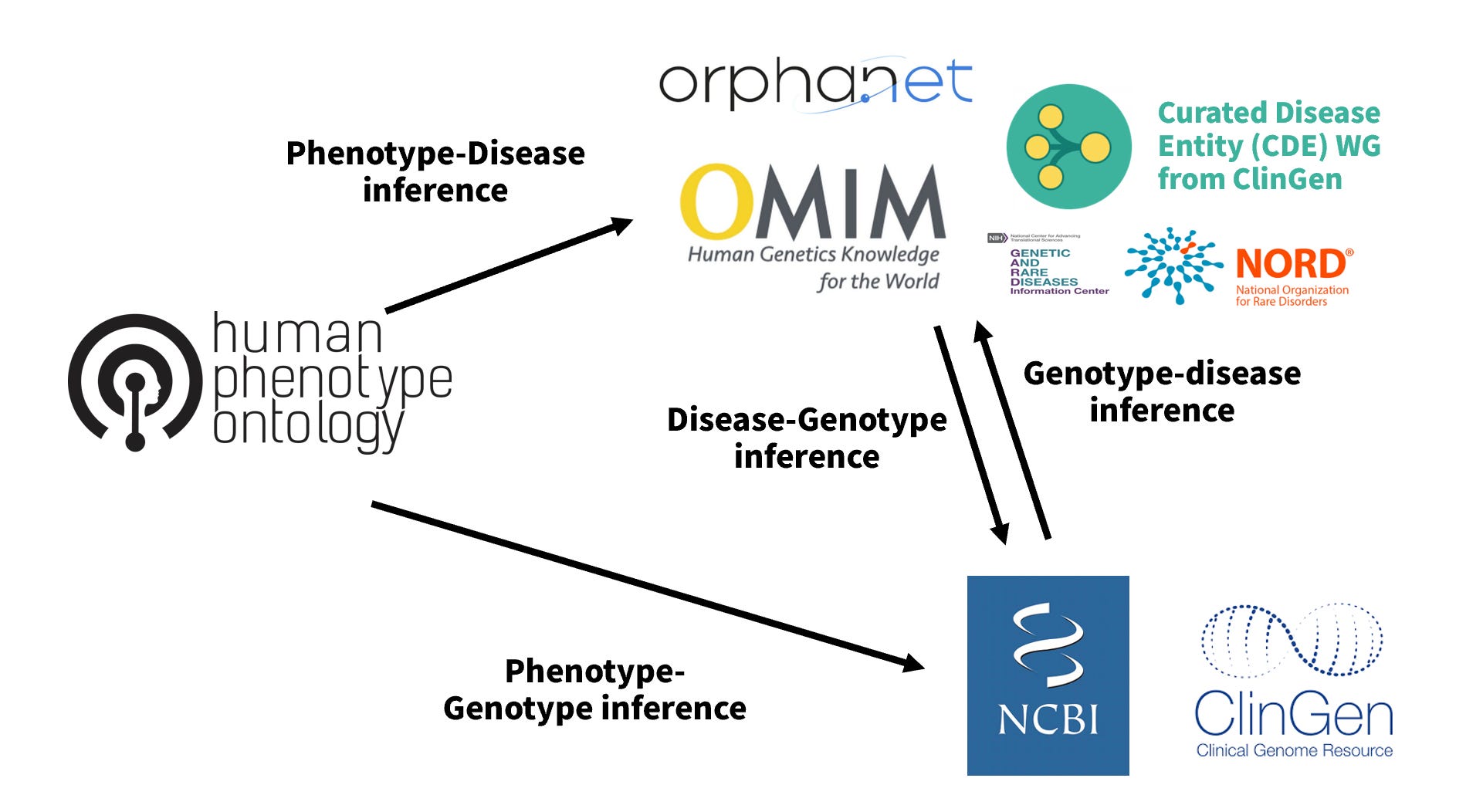
HPO 可以輸入單個表徵(Phenotype)進行檢索,適合用在單系統疾病,但對於多系統疾病,HPO 目前還無法輸入多個表徵然後羅列相關的疾病與基因。我便用 Python 串接 HPO 的 API,將多個疾病與基因候選抓出來並依相符表徵個數排列,Repo “hpo-many-phenotypes-query“ 放在 github 上。
切記表徵太混亂的還是需要人工介入,重複性高且需要穩定輸入輸出的就可以靠 API。
候選基因變異篩選定位(Candidate Genetic Variant Selections)
全外顯子定序(Whole exon sequencing, WES)就可能會抓到一個人數百個和人類參考基因有區別的變異,但不是所有的變異都是異常,有些會被視為是良性的「基因多型性(Genetic polymorphism)」。常見變異(common variants)通常定義上會抓 >10%,不過其實可以抓得更低到 > 1%。這些常見變異就不會被列入潛在致病的變異。
對於多系統罕病我的做法是對 HPO 進行 match & rank,也有些做法是從 gene pathway 著手(例如用 STRING),這塊我就比較不熟悉,有機會再多多學習。
致病力查詢(Pathogenicity Queries)
癌症可以靠查詢 COSMIC 與 OncoKB 資料庫,如果不限於癌症,大部份則使用 ClinVar。
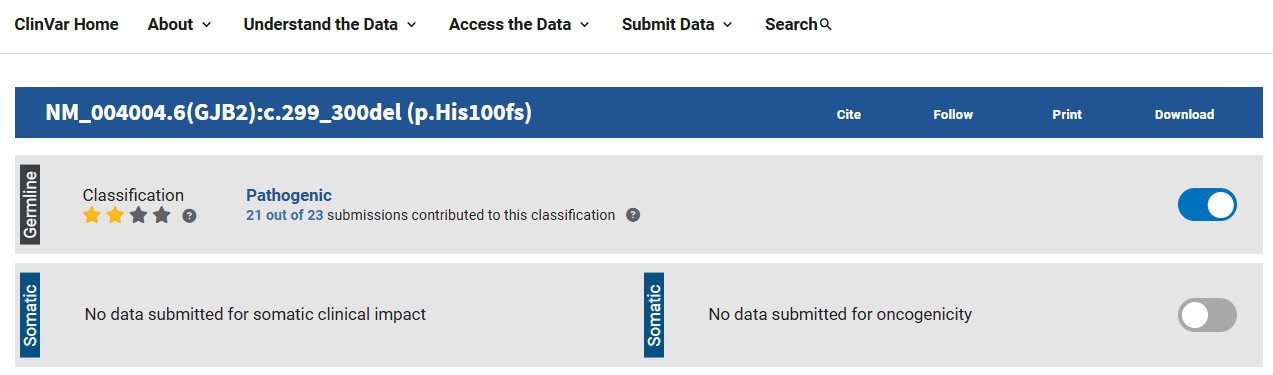
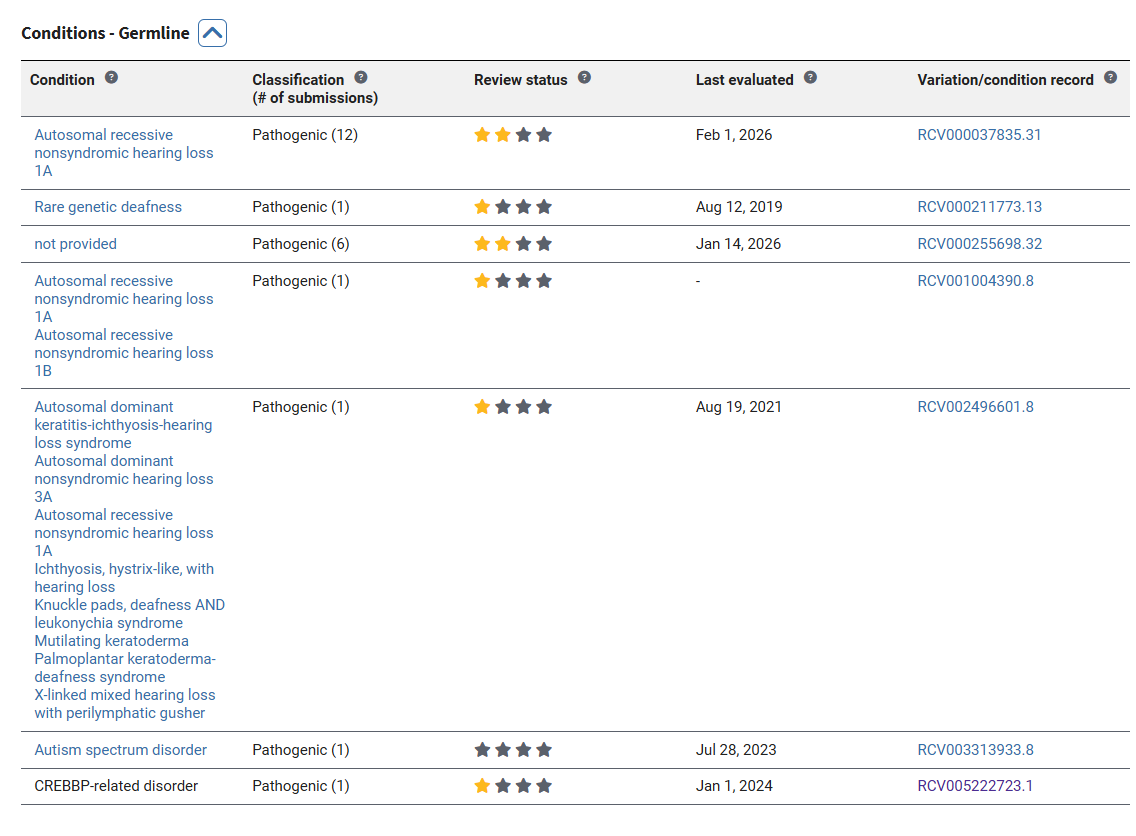
ClinVar 如果收錄該變異的話,則通常會提供致病性的分級(Five-point scale of variants):良性(benign)、可能良性(likely benign)、未確定(variants of uncertain significance, VUS)、可能致病(likely pathogenic)、致病(pathogenic)。
至於 ACMG 的致病力評級則有許許多多的規則指引,十分複雜,可以參考下圖。
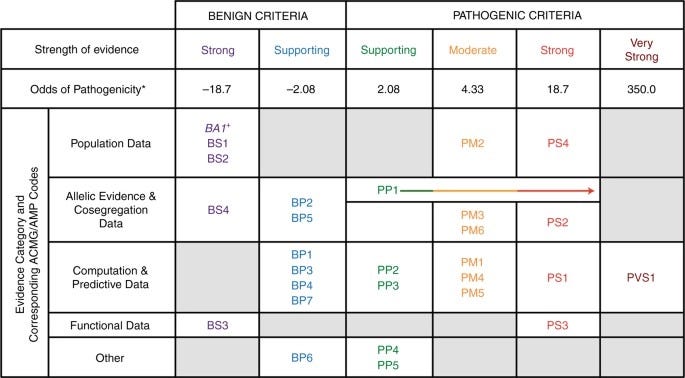
ACMG不像 ClinVar 只接搜詢到就給你一個評級。ClinVar 的壞處是沒收錄到或著是 VUS,就需要另請高明找 ACMG。ACMG 從理解到摸熟還需要一些時間。
臨床意義未明的變異評估(Variants of Uncertain Significance Evaluation)
如果 ClinVar 評為 VUS 或者是沒有收錄的,就會在這邊。這一塊也是我認為最複雜,生資方面最百家爭鳴的一塊。它的精髓在於預測基因組變異(Variant Effect Prediction, VEP)對基因、蛋白質產物或調控區的潛在功能影響與分子後果。
所有的突變可以先透過 VariantValidator 確認「座標系」,它會提供有點像是基因身份證號的「檢索號碼(Accession number)」。確認沒有語法錯誤後再丟到綜合查詢介面像 SpliceAI Lookup 與 Varsome 中看是否有各項我們電腦的基因組變異預測(in silico VEP)。
接下來便是對眾多變異分而擊之(divide and conquer)。我會將變異先區分為「錯義突變(missense mutation)」、「插入缺失突變(indel mutation)」。錯義突變有豐富的工具可供預測,我比較常用的是 Google 的 AlphaMissense、REVEL、CADD、PhyloP 與 SpliceAI。每種預測模型的核心不太一樣,AlphaMissense 從 AlphaFold 衍生概念,運用 protein language models,REVEL 收羅了各種傳統的錯義變異預測工具、CADD 整合多種基因標註資料,並對破壞性(deleteriousness)進行評估;PhyloP 則對跨物種演化保守性進行評估,越保守通常表示一旦有突變,功能被毀壞的機率越大;SpliceAI 則是看這個變異是否會造成剪切的異常(Abberrant splicing)。
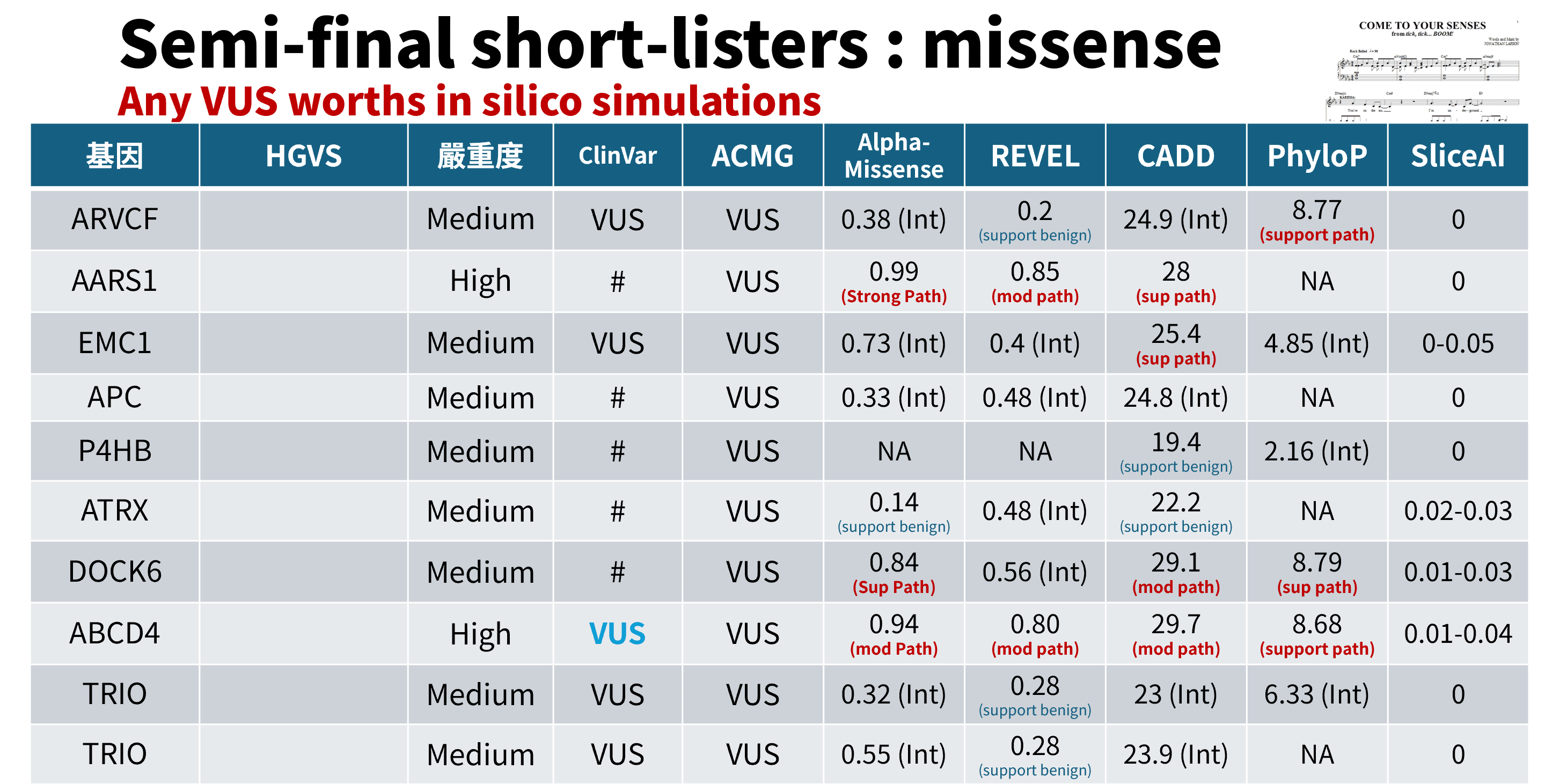
這個表則可以看出不同的預測模型也會產生不太一樣的結果。如果有多種統計方法預測該變異會對基因或基因產物造成有害的影響,包括保守性預測、演化預測、剪接位點影響等,就可以符合 ACMG 的 PP3: 電腦預測證據(in silico evidence)。
接下來則是比較麻煩的部份。Indels 可以進一步再分為發生在「密碼子(Codon)」與「非密碼子(Noncodon)」的突變。如果出現 frameshift 則要非常注意,看是否遇到提早終止(Early Termination)。如果有的話,則要到蛋白質資料庫如 UniProt 進行檢索,比對受影響的 domain 與結構,看是否出現 loss-of-function mutation(LoF)以符合 PVS1 rule。
Codon indels 的 VEP 我目前只用了 PROVEAN 與 PhyloP 和 SpliceAI。PROVEAN 是我主要的參考模型,JVI 已經將網頁版下架,目前僅剩 Linux 可以跑,我也藉由這個過程摸了一回 Linux 語法、Command line interface,以上與 PROVEAN 奮鬥的過程可以再寫一篇文章。
Non-codon indels 我則用 AlphaGenome 來進行預測,本站也有文章簡介其設計邏輯與RNA 剪接預測能力。


AlphaGenome 的使用法則可以參照我這個 repo “alphagenome-noncodon-indel-effect-prediction“ 進行。判讀的細節我還在摸索當中,之後有機會再分享進一步的心得。
基因型與表型對照(Genotype-Phenotype Mapping)
歷經上個階段辛苦的歷程,接下來則是將以下幾個變異整理出來:
ClinVar 登記為 likely pathogenic (LP) 與 pathogenic (P)
蛋白質結構與功能資料庫顯示為功能喪失變異(LoF)
多個 in silico VEP 顯示 LP/P
再來用這些變異的基因查詢 OMIM 並比對病人表型,並進行該基因的文獻搜詢以瞭解其功能和變異後可能的疾病表現。這個流程我還沒能好好地自動化,它大概也需要不少醫學上的背景知識才有辦法做得更好。
總結
次世代定序報告出來後,還是有一大半的病人無法找到致病基因,有些甚至連疾病名稱還不清楚,只有多個臨床表徵的鬆散集合。透過更嚴謹的生資分析、更新變異資料、重整臨床表型,在一些研究中甚至高達 70% 未被診斷的病人可以得到好的診斷。在這個時代,我們不缺講著「會用 AI 的人打敗不會用的人」這種無關痛癢的話,我們需要的是更多想要釐清問題並著手拆解的人。為此,我非常感謝林口長庚檢驗醫學部的楊醫師、余醫師、溫醫師與闕主任,這些醫生都是真心想解決臨床問題的人。